معرفی bioinformatics :
بیوانفورماتیک علم نوینی است که در آن با استفاده از کامپیوتر، نرم افزارهای کامپیوتری و بانکهای اطلاعاتی سعی میگردد تا به مسائل بیولوژیکی بخصوص در زمینه های سلولی و ملکولی پاسخ داده شود در این علم با بکارگیری کامپیوتر سعی می گردد تا تحقیقات وسیعتری در خصوص پروتئینها و ژنها بعمل آید.
در چند دههٔ اخیر، پیشرفت در زیستشناسی مولکولی و تجهیزات مورد نیاز تحقیق در این زمینه باعث افزایش سریع تعیین توالی ژنوم بسیاری از گونههای موجودات شد، تا جایی که پروژههای تعیین توالی ژنومها از پروژههای بسیار رایج به حسب میآیند. امروزه توالی ژنوم بسیاری از موجودات ساده مانند باکتریها تا موجودات بسیار پیشرفته چون یوکاریوتهای پیچیده شناسایی شدهاست. پروژهٔ شناسایی ژنوم انسان در سال ۱۹۹۰ آغاز شد و در سال ۲۰۰۳ پایان یافت و اکنون اطلاعات کامل مربوط به توالی هر ۲۴ کروموزوم انسان موجود است.
معرفی BLAST :
در علوم سلولی و مولکولی و ژنتیک (بیو انفورماتیک) ، BLAST یک ابزار پایهای برای جستجوی تطبیقهای موضعی یک لگاریتم کاربردی برای مقایسه اطلاعات یا ( سکانس ) بیولوژیک می باشد. با این نرم افزار می توان سکانس اسید های آمینه در پروتئین ها یا نوکلئوتید ها را در DNA را با هم مقایسه کرد . این نرم افزار به پژوهشگر اجازه می دهد تا یک سکانس را با سکانس مرجع یا سکانسی که در data base وجود دارد ، مقایسه کرد . شناسایی سکانس های موجود در data base که بیشترین شباهت را با سکانس مورد نظر دارد از دیگر قابلیت های این نرم افزار است . بر حسب نوع سکانس انواع مختلفی از بلاست امکان پذیر است . مثلا اگر یک ژن ناشناخته در موش که قبلا اطلاعاتی از آن در اختیار نبوده ، باید بررسی شود ، یک محقق ترجیح می دهد این سکانس را با ژنوم انسان بلاست کند. این نرم افزار درNIH ( موسسه ملی بهداشت آمریکا) طراحی شد . بلاست یکی از برکاربردترین نرم افزارها در بیوانفورماتیک است که با سرعت مطلوب مقایسه مورد نظر را انجام می دهد . سرعت زمانی اهمیت خود را نشان می دهد که با ژنوم کامل روبرو باشیم . پیش از طراحی این نرم افزار مقایسه سکانس ها بسیار وقت گیر بود .
از blast در موارد زیر استفاده می شود:
- کدام گونه ها ی باکتریایی دارای پروتئین خاصی هستند که با یک پروتین که سکانس آن مشخص است ، شباهت دارد
- یک سكانس خاص DNA از کجا منشا گرفته است .
- چه ژنهایی یک موتیف یا ساختار ویژه را دارند .
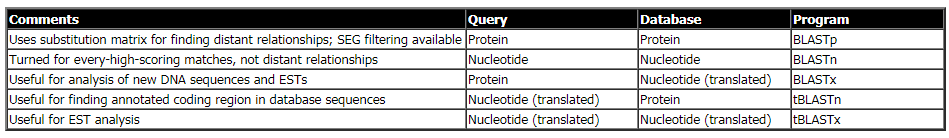
انواع BLAST :
بنا به نوع توالی مورد نظر و نوع پایگاه مورد جستجو، برنامه های BLAST طراحی شده اند که در زیر توضیح داده می شوند. البته برای هر یک از این برنامه ها نیز زیربرنامه هایی که در آن تنظیمات بهینه شده است معرفی شده اند و مانند BLASTN برای توالی کوتاه.
BLASTN :
در این نوع بلست، توالی مورد تقاضا نوکلئوتیدی است و جستجو در پایگاه توالی های نوکلئوتیدی انجام می شود. نتیجه جستجو جفت توالی های نوکلئوتیدی مشابه است که براساس شاخص های آماری میزان شباهت و یکسانی آن نشان داده می شود.
BLASTP :
در این نوع BLAST، توالی مورد تقاضا پروتئینی است و جستجو در پایگاه توالی های پروتئینی انجام می شود. نتیجه جستجو توالی های پروتئینی مشابه به توالی الگو است که بر اساس شاخص های آماری میزان شباهت و یکسانی آن ها با توالی الگو نشان داده می شود.
BLASTX :
در این نوع، توالی مورد تقاضا نوکلئوتیدی است که در 6 قالب خواندی (ORF) ترجمه شده و به صورت توالی پروتئینی در پایگاه توالی های پروتئینی جستجو می شود. نتیجه جستجو توالی های مشابه با توالی الگو است که براساس آن میتوانیم به توالی جدید خود قالب خواندنی و عملکرد نسبت دهیم.
tBLASTN :
در این نوع BLAST، توالی مورد تقاضا پروتئینی است و جستجو در پایگاه توالی های نوکلئوتیدی انجام می شود که در 6 قالب خواندنی ترجمه شده است. نتیجه جستجو توالی های مشابه با توالی مورد تقاضاست که براساس آن می توانیم برای توالی پروتئینی خود توالی های رمز کننده ی آن را شناسایی کنیم.
tBLASTX :
در این نوع بلست، توالی مورد تقاضا نوکلئوتیدی است که در 6 قالب خواندنی به پروتئین ترجمه می شود و در پایگاه توالی های نوکلئوتیدی که آن نیز در 6 قالب خواندی به پروتئین ترجمه می شود مورد جستحو قرار می گیرد. این نوع جستجو بویژه در مطالعات EST به کار می رود.
الگوریتم :
برای اجرای بلاست نیاز به دو دنباله میباشد یکی دنبالهٔ درخواستی یا مورد نظر و دیگری دنبالهٔ هدف یا دنبالههای موجود در پایگاه دادهای از دنبالهها. بلاست زیر دنبالههایی از پایگاه داده را پیدا میکند که شبیه دنبالهٔ ورودی (query) باشند. معمولاً دنبالهٔ query بسیار کوچکتر از پایگاه داده است. به عنوان مثال query ممکن است هزار نوکلئوتیدی باشد در حالی که پایگاه داده از چندین بیلیون نوکلئوتید تشکیل شده باشد. ایدهٔ اصلی الگوریتم بلاست این است که به دنبال تطابقهای با بیشترین امتیاز بین query و پایگاه داده میگردد بر اساس تقریب زدن یک الگوریتم اکتشافی به اسم Smith-Waterman_algorithm. الگوریتم Smith-Waterman بسیار زمانگیر است از اینرو در بلاست از یک روش اکتشافی (heuristic) که البته دقت کمتری خواهد داشت استفاده میشود. اما در عوض ۵۰ بار سریعتر است. در زیر مراحل الگوریتم بلاست (پروتئین به پروتئین) به صورت خلاصه آورده شده است:
- حذف قسمتهای تکراری از دنبالهٔ query (قسمتهای با پیچیدگی کم): قسمتهای با پیچیدگی کم قسمتهایی در دنباله هستند که عناصرشان تنوع کمی دارند.
- ساخت یک لیست k حرفی از دنباله query:
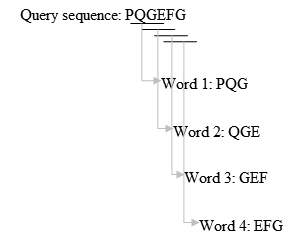
1- لیست کردن تطابقهای ممکن:
این مرحله یکی از تفاوتهای اساسی بین بلاست و FASTA است. برای FASTA تمام لغات مشترک در پایگاه داده و دنبالههای کوئری ای که در مرحلهٔ دوم لیست شدند مهم است، اما برای بلاست فقط لغات با امتیاز بالا اهمیت دارند.
1-سازماندهی لغات باقیمانده (لغات با امتیاز بیشتر از حد آستانه) و تبدیل آن به یک درخت جستجوی بهینه:
هدف این قسمت این است که برنامه بتواند سریعتر لغات با امتیاز بالا را با دنبالههای پایگاه داده مقایسه کند.
- تکرار مراحل ۳ و ۴ به ازای هر لغت k-حرفی در دنباله query.
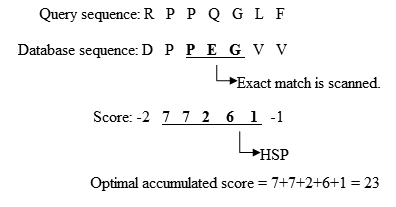
- جستجو در پایگاه داده برای پیدا کردن تطابق دقیق بین دنبالههای باقیمانده و دنبالههای پایگاه داده:
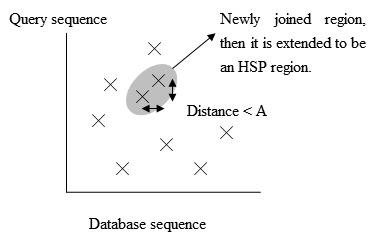
- گسترش تطابقهای دقیق به HSP (جفت دنبالههای با امتیاز تطابق بالاتر از حد آستانه)
- بررسی معناداری امتیاز داده شده به HSPها.
در این مرحله، بلاست میزان معناداری آماری امتیاز داده شده به هر HSP را با استفاده از Gumbel extreme value distribution (EVD) بررسی میکند. بر طبق Gumbel EVD احتمال اینکه با احتمال p امتیاز مشاهده شود مثل S که بزرگتر یا مساوی x باشد برابر است با:

به طوریکه:
![{\displaystyle \mu ={}^{\left[\log \left(Km'n'\right)\right]}\!\!\diagup \!\!{}_{\lambda }\;}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f345aa27fbc08d03c73a72bd3581e3bd26f6a7ca)
- ادغام HSPها و تبدیل آنها به تطابقهای بزرگتر
- نمایش تطابق محلی دنباله query با پایگاه داده با استفاده از الگوریتم gapped Smith-Waterman.
- انتخاب تطابقهایی که مقدار مورد انتظار امتیاز آنها کمتر از حد آستانهٔ E شده باشد.

-----------------------------------------------
منابع : سایت ژنتیک ، ویکی پدیا , سایت ncbi ، پرتال بیوانفورماتیک