در اینجا کدهای اساسی و الگو مورد استفاده در بسیاری از شبیه سازی های سیستم های فیزیکی با ارائه ی توضیحات اجمالی و مختصر ارائه شده است و میتوانید شرح تکمیلی هر بخش را در قسمت آموزش نرم افزار لمپس مشاهده نمایید.
کد شماره 1:
کد ورودی مربوط به شبیه سازی یک ساختار ساده مانند آلومینیوم و انجام فرایند بهینه سازی و یافتن ثابت شبکه متناسب با بهینه سازی انرژی مربوطه.
# Find minimum energy fcc configuration
# Mark Tschopp, 2010
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ---------- Create Atoms ---------------------
lattice fcc 4
region box block 0 1 0 1 0 1 units lattice
create_box 1 box
lattice fcc 4 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 box
replicate 1 1 1
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
# ---------- Run Minimization ---------------------
reset_timestep 0
fix 1 all box/relax iso 0.0 vmax 0.001
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
min_style cg
minimize 1e-25 1e-25 5000 10000
variable natoms equal "count(all)"
variable teng equal "c_eatoms"
variable length equal "lx"
variable ecoh equal "v_teng/v_natoms"
print "Total energy (eV) = ${teng};"
print "Number of atoms = ${natoms};"
print "Lattice constant (Angstoms) = ${length};"
print "Cohesive energy (eV) = ${ecoh};"
print "All done!"
بخشهای مختلف این کد به شرح زیر است:
الف- آماده سازی محیط شبیه سازی
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
ب- تعریف هندسه و ساختار اولیه ماده ی موردنظر (آلومینیوم)
# ---------- Create Atoms ---------------------
lattice fcc 4
region box block 0 1 0 1 0 1 units lattice
create_box 1 box
lattice fcc 4 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 box
replicate 1 1 1
پ- تعریف پتانسیل موجود در بین اتم های ساختار شبیه سازی شده (فورس فیلد)
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
ت- انجام محاسبات موردنظر در شبیه سازی (ازقبیل محاسبه ی انرژی و ...)
# ---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
ث- انجام فرایند بهینه سازی انرژی بر روی ساختارشبیه سازی شده ی اولیه
# ---------- Run Minimization ---------------------
reset_timestep 0
fix 1 all box/relax iso 0.0 vmax 0.001
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
min_style cg
minimize 1e-25 1e-25 5000 10000
ج- نامگذاری خروجی های حاصل از شبیه سازی (تعریف متغییر)
variable natoms equal "count(all)"
variable teng equal "c_eatoms"
variable length equal "lx"
variable ecoh equal "v_teng/v_natoms"
چ- نمایش و پرینت خروجی ها
print "Total energy (eV) = ${teng};"
print "Number of atoms = ${natoms};"
print "Lattice constant (Angstoms) = ${length};"
print "Cohesive energy (eV) = ${ecoh};"
print "All done!"
کد شماره 2:
# Find minimum energy fcc configuration
# Mark Tschopp, 2010
# This requires the variable latconst to be input via the command line
# e.g., lmp_win_no-mpi -var latconst 4 < calc_fcc_ver1.in
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ---------- Create Atoms ---------------------
lattice fcc ${latconst}
region box block 0 1 0 1 0 1 units lattice
create_box 1 box
lattice fcc ${latconst} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 box
replicate 1 1 1
# ---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
# ---------- Run Minimization ---------------------
reset_timestep 0
fix 1 all box/relax iso 0.0 vmax 0.001
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
min_style cg
minimize 1e-25 1e-25 5000 10000
variable natoms equal "count(all)"
variable teng equal "c_eatoms"
variable length equal "lx"
variable ecoh equal "v_teng/v_natoms"
print "Total energy (eV) = ${teng};"
print "Number of atoms = ${natoms};"
print "Lattice constant (Angstoms) = ${length};"
print "Cohesive energy (eV) = ${ecoh};"
print "All done!"
کد ورودی مربوط به شبیه سازی یک ساختار آلمینیوم و پیدا کردن کمترین انرژی برای این ساختار.
در این کد و در قسمتinitilize simulation اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت create atms اتم ها ساخته می شوند. در قسمت define interatomic potential پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت define settings نیز تنظیمات موردنظر خود ازقبیل تعیین محاسبات موردنظر را ایجاد کرده و در قسمت پایانی یعنی run minimization فرایند یافتن کمترین انرژی ساختار شبیه سازی شده با لمپس انجام می شود.
کد شماره 3:
# Input file for uniaxial tensile loading of single crystal aluminum
# Mark Tschopp, November 2010
# ------------------------ INITIALIZATION ----------------------------
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 4.05
# ----------------------- ATOM DEFINITION ----------------------------
lattice fcc ${latparam}
region whole block 0 10 0 10 0 10
create_box 1 whole
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region whole
# ------------------------ FORCE FIELDS ------------------------------
pair_style eam/alloy
pair_coeff * * Al99.eam.alloy Al
# ------------------------- SETTINGS ---------------------------------
compute csym all centro/atom fcc
compute peratom all pe/atom
######################################
# EQUILIBRATION
reset_timestep 0
timestep 0.001
velocity all create 300 12345 mom yes rot no
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
# Set thermo output
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
# Run for at least 10 picosecond (assuming 1 fs timestep)
run 20000
unfix 1
# Store final cell length for strain calculations
variable tmp equal "lx"
variable L0 equal ${tmp}
print "Initial Length, L0: ${L0}"
######################################
# DEFORMATION
reset_timestep 0
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1
variable srate equal 1.0e10
variable srate1 equal "v_srate / 1.0e12"
fix 2 all deform 1 x erate ${srate1} units box remap x
# Output strain and stress info to file
# for units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa]
# p2, p3, p4 are in GPa
variable strain equal "(lx - v_L0)/v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_SC_100.def1.txt screen no
# Use cfg for AtomEye
dump 1 all cfg 250 dump.tensile_*.cfg mass type xs ys zs c_csym c_peratom fx fy fz
dump_modify 1 element Al
# Display thermo
thermo 1000
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press
run 20000
######################################
# SIMULATION DONE
print "All done"
کد ورودی مربوط به اعمال تنش (کشش) به ساختار کریستالی آلومینیوم و بررسی نمودار تنش- کرنش حاصله.
در این کد و در قسمت initilization اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت atom definition اتم ها ساخته می شوند. در قسمت force fields پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت settings نیز تنظیمات موردنظر خود ازقبیل ایجاد تعادل در سیستم و ایجاد تنش کرده و در قسمت پایانی یعنی simulation done پیام انجام شبیه سازی را چاپ می کنیم.
کد شماره 4:
کد ورودی مربوط به اعمال تنش (تراکم) به ساختار کریستالی آلومینیوم و بررسی نمودار تنش- کرنش حاصله.
# Input file for uniaxial compressive loading of single crystal aluminum # Mark Tschopp, November 2010 # ------------------------ INITIALIZATION ---------------------------- units metal dimension 3 boundary p p p atom_style atomic variable latparam equal 4.05 # ----------------------- ATOM DEFINITION ---------------------------- lattice fcc ${latparam} region whole block 0 10 0 10 0 10 create_box 1 whole region upper block INF INF INF INF INF INF units box lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1 create_atoms 1 region upper # ------------------------ FORCE FIELDS ------------------------------ pair_style eam/alloy pair_coeff * * Al99.eam.alloy Al # ------------------------- SETTINGS --------------------------------- compute csym all centro/atom fcc compute peratom all pe/atom ###################################### # EQUILIBRATION reset_timestep 0 timestep 0.001 velocity all create 300 12345 mom yes rot no fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1 # Set thermo output thermo 1000 thermo_style custom step lx ly lz press pxx pyy pzz pe temp # Run for at least 10 picosecond (assuming 1 fs timestep) run 20000 unfix 1 # Store final cell length for strain calculations variable tmp equal "lx" variable L0 equal ${tmp} print "Initial Length, L0: ${L0}" ###################################### # DEFORMATION reset_timestep 0 fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1 variable srate equal 1.0e10 variable srate1 equal "-v_srate / 1.0e12" fix 2 all deform 1 x erate ${srate1} units box remap x # Output strain and stress info to file # for units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa] # p2, p3, p4 are in GPa variable strain equal "(lx - v_L0)/v_L0" variable p1 equal "v_strain" variable p2 equal "-pxx/10000" variable p3 equal "-pyy/10000" variable p4 equal "-pzz/10000" fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_comp_100.def1.txt screen no # Use cfg for AtomEye dump 1 all cfg 250 dump.comp_*.cfg mass type xs ys zs c_csym c_peratom fx fy fz dump_modify 1 element Al # Display thermo thermo 1000 thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press run 20000 ###################################### # SIMULATION DONE print "All done"
در این کد و در قسمت initilization اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت atom definition اتم ها ساخته می شوند. در قسمت force fields پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت define settings نیز تنظیمات موردنظر خود ازقبیل ایجاد تعادل در سیستم و ایجاد تنش کرده و در قسمت پایانی یعنی simulation done پیام انجام شبیه سازی را چاپ می کنیم.
کد شماره 5:
کد ورودی مربوط به انجام محاسبات تنش لازم برای از بین بردن تقارن ساختاری آهن.
############################################################################
# Interfacial Fracture
# Mark Tschopp, Nathan Rhodes 2011
# lmp_exe -var datfile Fe_110_sig3.txt -var nloop 100 < in.gb_fracture.txt
# Simulation deletes atoms outside of +/- deldist from GB and constrains and pulls
# atoms outside of +/- fixdist from GB to fracture the GB
############################################################################
#variable datfile index Fe_110_sig3.txt
variable strain equal 0.001
#variable nloop equal 100
variable repx equal 1
variable repz equal 1
variable strain2 equal "1+v_strain"
variable deldist equal 50
variable fixdist equal 45
######################################
# INITIALIZATION
units metal
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
######################################
# SIMULATION CELL VARIABLES (in Angstroms)
read_data ${datfile}
#variable minlength equal 100
variable xlen equal lx
variable ylen equal ly
variable zlen equal lz
print "lx: ${xlen}"
print "ly: ${ylen}"
print "lz: ${zlen}"
# Replicate simulation cell in each direction
replicate ${repx} 1 ${repz}
######################################
# INTERATOMIC POTENTIAL
pair_style eam/fs
pair_coeff * * Fe-C_Hepburn_Ackland.eam.fs Fe C
# Compute stress information for Atomeye visualization
compute stress all stress/atom
compute stress1 all reduce sum c_stress[1]
compute stress2 all reduce sum c_stress[2]
compute stress3 all reduce sum c_stress[3]
compute stress4 all reduce sum c_stress[4]
compute stress5 all reduce sum c_stress[5]
compute stress6 all reduce sum c_stress[6]
##########################################
# Minimize first
reset_timestep 0
thermo 10
thermo_style custom step lx ly lz press pxx pyy pzz pe c_stress1 c_stress2 c_stress3 c_stress4 c_stress5 c_stress6
min_style cg
fix 1 all box/relax x 0.0 z 0.0 couple none vmax 0.001
minimize 1.0e-25 1.0e-25 1000 10000
unfix 1
# Compute distance for each side of the grain boundary to displace
variable ly1 equal ly
variable ly0 equal ${ly1}
variable lydelta equal "v_strain*v_ly0/2"
# Setup file output (time in ps, pressure in GPa)
variable p1 equal "(ly-v_ly0)/v_ly0"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
variable p5 equal "-pxy/10000"
variable p6 equal "-pxz/10000"
variable p7 equal "-pyz/10000"
variable p8 equal "pe"
# Output stress and strain information to datafile for Matlab post-processing
fix equil1 all print 1 "${p1} ${p2} ${p3} ${p4} ${p5} ${p6} ${p7} ${p8}" file data.${datfile}.txt screen no
fix 1 all nve
run 1
unfix 1
variable pressf1 equal pyy
variable pressf equal ${pressf1}
##########################################
# Create cfg files with stress in y direction for AtomEye viewing
dump 1 all cfg 500 ${datfile}_*.cfg id type xs ys zs c_stress[2]
##########################################
# CREATE REGIONS FOR BOUNDARY CONDITIONS
# Delete groups of atoms far from boundary
region rlow block 0 200 -200 -${deldist} 0 200 units box
region rhigh block 0 200 ${deldist} 200 0 200 units box
group glow region rlow
group ghigh region rhigh
delete_atoms group glow
delete_atoms group ghigh
# Create groups to fix and displace
region rgblow block 0 200 -200 -${fixdist} 0 200 units box
region rgbhigh block 0 200 ${fixdist} 200 0 200 units box
group gbhigh region rgbhigh
group gblow region rgblow
# Put fixed boundary condition on edge atoms by setting forces to zero
fix 2 gbhigh setforce 0 0 0
fix 3 gblow setforce 0 0 0
##########################################
# MS Deformation loop
variable a loop ${nloop}
label loop
# Increase box bound and minimize again
#reset_timestep 0
displace_box gblow y delta -${lydelta} 0 units box
displace_box gbhigh y delta 0 ${lydelta} units box
minimize 1.0e-25 1.0e-25 1000 10000
run 1
print "Pressf: ${pressf}"
variable pdiff equal "pyy - v_pressf"
print "Pressf: ${pressf}"
print "Pdiff: ${pdiff}"
#if ${pdiff} > 10000 then "exit"
variable pressf1 equal pyy
variable pressf equal ${pressf1}
next a
jump in.gb_fracture.txt loop
unfix equil1
######################################
# SIMULATION DONE
print "All done"
در این کد و در قسمت initilize اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت simulation cell variables متغیرهای مربوط سلول اتم ها ساخته می شوند. در قسمت interatomic potential پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت minimize در ساختار شبیه سازی شده بهینه سازی انرژی صورت می گیرد. سپس در دوقسمت بعدی create region و MD deformation loop با تعریف ناحیه مرزی اقدام به ایجاد تغییر شکل و دیفرمیشن در ساختار می نماییم و در قسمت پایانی یعنی simulation done پیام انجام شبیه سازی را چاپ می کنیم.
کد شماره 6:
کد ورودی مربوط به تغییر شکل نانولوله در نرم افزار لمپس.
# Deforming a Nanowire.
# Suman Kandel, Dr. Mark A. Tschopp, 2011.
# ------------------------ INITIALIZATION ----------------------------
units metal
boundary p p p
atom_style atomic
# ----------------------- ATOM DEFINITION ----------------------------
lattice hcp 3.20
region whole block 0 100 0 100 0 100 units box
create_box 1 whole
region LLF cylinder z 50 50 20 INF INF units box
lattice hcp 3.20 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region LLF
#delete_atoms overlap 0.5 all all
# ------------------------ FORCE FIELDS ------------------------------
pair_style eam/fs
pair_coeff * * Al-Mg.eam.fs Mg
# ------------------------- SETTINGS ---------------------------------
#### Computes Required
compute csym all centro/atom 12
compute 2 all stress/atom
compute mytemp all temp
compute 11 all reduce sum c_2[1]
compute 12 all reduce sum c_2[2]
compute 13 all reduce sum c_2[3]
compute 14 all reduce sum c_2[4]
compute 15 all reduce sum c_2[5]
compute 16 all reduce sum c_2[6]
neighbor 0.3 bin
neigh_modify delay 10
thermo 100
velocity all create 100.0 16723
thermo_style custom step temp etotal press pxx pyy pzz lx ly lz
shell mkdir dump
shell cd dump
#---------------Energy Equilibriation--------------------------
reset_timestep 0
fix 1 all nve
fix 2 all temp/rescale 10 100.0 100.0 1.0 0.5
dump 1 all custom 10000 dump.equilibrate.* id type x y z
run 10000
unfix 1
unfix 2
#-----------------Pressure Equilibriation-----------------------
fix 1 all npt 100.0 100.0 10.0 aniso NULL NULL NULL NULL 0.0 0.0 10 drag 0.3
run 10000
unfix 1
undump 1
shell cd ..
shell mkdir deform
shell cd deform
#--------------------Storing Initial length---------------------
variable tmp equal "lz"
variable L0 equal ${tmp}
print "Initial Length, L0: ${L0}"
variable strain equal "v_srate/1e12"
#------------Deform------------------------------
reset_timestep 0
fix 1 all deform 1 z erate 0.0001 units box
fix 2 all nvt 100.0 100.0 10.0
variable strain equal "(lz - v_L0)/v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
variable p5 equal "lx"
variable p6 equal "ly"
variable p7 equal "lz"
variable p8 equal "temp"
variable p9 equal "pe"
variable p10 equal "ke"
variable p11 equal "-pxy/10000"
variable p13 equal "-pyz/10000"
variable p12 equal "-pxz/10000"
variable fm equal "(v_p2+v_p3+v_p4)/3" ##### Hydrostatic stress
variable fv equal "sqrt((v_p2-v_p3)^2+(v_p3-v_p4)^2+(v_p4-v_p2)^2+6*(v_p11^2+v_p12^2+v_p13^2)/2)" ######Von Mises Stress
variable t equal "v_fm/v_fv"
variable fd equal (((v_p2-v_fm)*(v_p3-v_fm)*(v_p4-v_fm))-v_p11^2*(v_p4-v_fm)-v_p12^2*(v_p3-v_fm)-v_p13^2*(v_p2-v_fm)+2*v_p11*v_p12*v_p13)####Deviatoric Von Mises stress
dump 2 all custom 10000 dump.defo.* id type x y z c_csym c_2[1] c_2[2] c_2[3] c_2[4] c_2[5] c_2[6]
fix def_print all print 100 "${p1} ${p2} ${p3} ${p4} ${p5} ${p6} ${p7} ${p8} ${p9} ${p10} ${p11} ${p12} ${p13} ${fm} ${fv} ${t} ${fd}" file mg001.defo.txt screen no
run 100000
در این کد و در قسمت initilizAtion اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت atom definition اتم ها ساخته می شوند. در قسمت force fields پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت settings نیز تنظیمات موردنظر خود ازقبیل ایجاد سرعت در اتم های موجود در شبیه سازی را اعمال می نماییم. در دوقسمت بعدی که شامل energy equilibriation و pressure equilibiration است متعادل سازی انرژی و فشار صورت پذیرفته و در گام بعدی storing initial lenght باذخیره سازی طول اولیه ساختار و انجام محاسبات دیگر با استفاده از دستور compute شبیه سازی را به پایان می بریم.
کد شماره 7:
کد ورودی مربوط به شبیه سازی زنجیره پلیمری در نرم افزار لمپس با استفاده از روش درشت دانه سازی.
#####################################################
# #
# #
# Filename: in.deform.polychain.txt #
# Author: Mark Tschopp, 2010 #
# #
# The methodology outlined here follows that from #
# Hossain, Tschopp, et al. 2010, Polymer. Please #
# cite accordingly. The following script requires #
# a LAMMPS data file containing the coordinates and #
# appropriate bond/angle/dihedral lists for each #
# united atom. #
# #
# Execute the script through: #
# lmp_exe < in.deform.polychain.txt #
# #
#####################################################
# VARIABLES
variable fname index data.txt
variable simname index PE_cl100
# Initialization
units real
boundary f f f
atom_style molecular
log log.${simname}.txt
read_data ${fname}
# Dreiding potential information
neighbor 0.4 bin
neigh_modify every 10 one 10000
bond_style harmonic
bond_coeff 1 350 1.53
angle_style harmonic
angle_coeff 1 60 109.5
dihedral_style multi/harmonic
dihedral_coeff 1 1.73 -4.49 0.776 6.99 0.0
pair_style lj/cut 10.5
pair_coeff 1 1 0.112 4.01 10.5
compute csym all centro/atom fcc
compute peratom all pe/atom
#####################################################
# Equilibration (Langevin dynamics at 5000 K)
velocity all create 5000.0 1231
fix 1 all nve/limit 0.05
fix 2 all langevin 5000.0 5000.0 10.0 904297
thermo_style custom step temp
thermo 10000
timestep 1
run 1000000
unfix 1
unfix 2
write_restart restart.${simname}.dreiding1
#####################################################
# Define Settings
compute eng all pe/atom
compute eatoms all reduce sum c_eng
#####################################################
# Minimization
dump 1 all cfg 6 dump.comp_*.cfg mass type xs ys zs c_csym c_peratom fx fy fz
reset_timestep 0
fix 1 all nvt temp 500.0 500.0 100.0
thermo 20
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
min_style cg
minimize 1e-25 1e-25 500000 1000000
print "All done"
در این کد در ابتدای کد و قسمت variables اقدام به تعریف متغییرها نموده و در قسمت initilize اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت dreiding potential پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و با دستورهای مریوط به بخش equilibration متعادل سازی سیستم صورت می پذیرد. در قسمت define settings نیز تنظیمات موردنظر صورت گرفته و در قسمت پایانی یعنی minimization بهینه سازی انرژی ساختار را انجام می دهیم.
کد شماره 8:
کد ورودی مربوط به شبیه سازی ساختار دو لایه ای نیکل و الومینیوم.
# Input file for bi-layered crystal, Nickle and Aluminum, relaxed
# Tikelia Wright, July 2012
# ------------------------ INITIALIZATION ----------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# ----------------------- ATOM DEFINITION -----------------------
lattice fcc 3.52
region upper block 0 54 0 54 0 7
lattice fcc 4.05
region lower block 0 47 0 47 6.5 13
region whole union 2 upper lower
create_box 2 whole
lattice fcc 3.52 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region upper
lattice fcc 4.05 origin 0 0 .247 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 2 region lower
# ------------------------ FORCE FIELDS -----------------------
pair_style eam/alloy
pair_coeff * * Mishin-Ni-Al-2009.eam.alloy Ni Al
#---------------------------Settings----------------------------
compute csym all centro/atom fcc
compute eng all pe/atom
compute eatoms all reduce sum c_eng
compute strs all stress/atom
#----------------------Run Minimization-------------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1 all custom 10 dump.comp.* id type xs ys zs c_csym c_eng c_strs[1] c_strs[2] c_strs[3]
min_style cg
minimize 1e-15 1e-15 5000 5000
######################################
# SIMULATION DONE
print "All done"
در این کد و در قسمت initilization اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت atom definition اتم ها ساخته می شوند. در قسمت force fields پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت run minimization فرایند بهینه سازی انرژی صورت گرفته و در قسمت پایانی یعنی simulation done پیام انجام شبیه سازی را چاپ می کنیم.
کد شماره 9:
کد ورودی مربوط به محاسبه ی انرژی تشکیل نقص تهی جایی در ساختار یک سیستم.
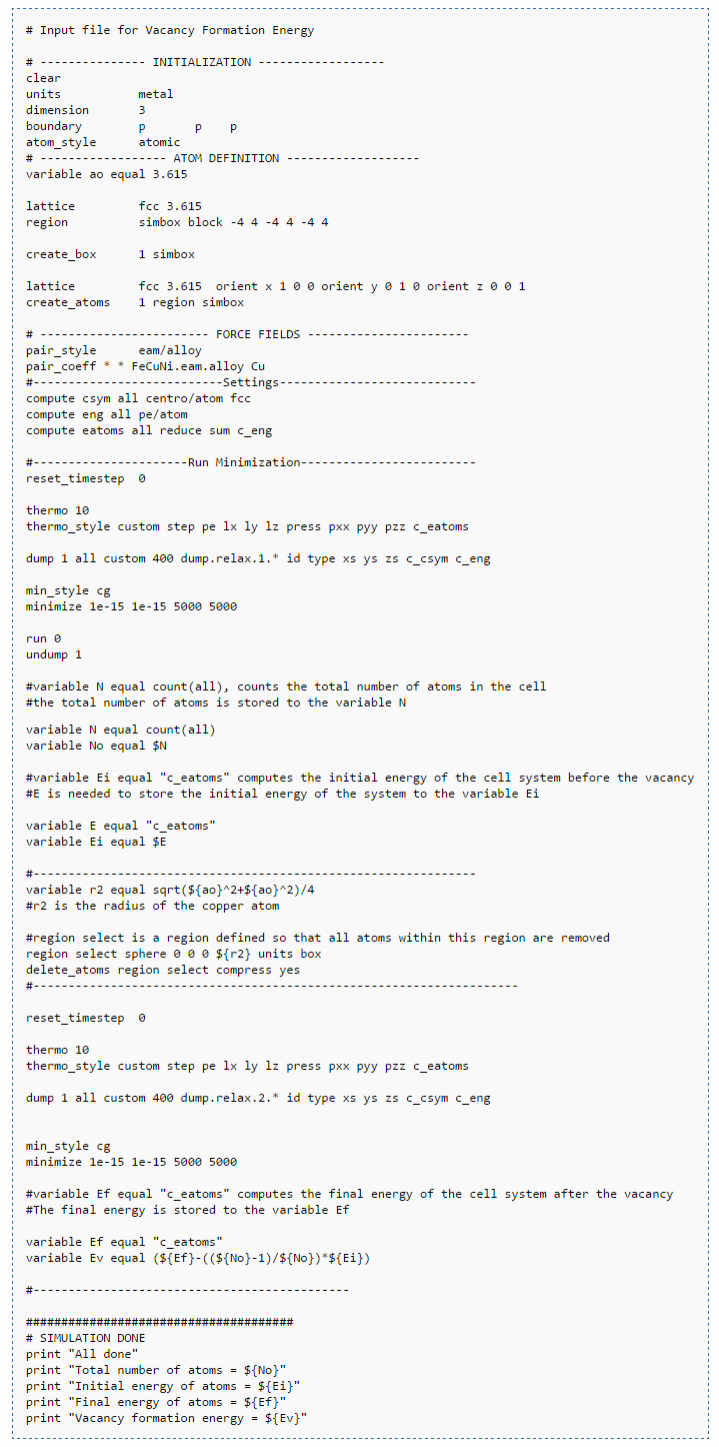
در این کد و در قسمت initilization اقدام به فراهم نمودن شرایط اولیه سیستم می کنیم سپس در قسمت atom definition اتم ها ساخته می شوند. در قسمت force fields پتانسیل و یا فورس فیلد مورد استفاده در شبیه سازی دینامیک مولکولی تعیین شده و در قسمت run minimization فرایند بهینه سازی انرژی صورت گرفته و در قسمت پایانی یعنی simulation done پیام انجام شبیه سازی را چاپ می کنیم.
------------------------------------
منبع : lammps.ir